En atención a la creciente preocupación sobre la confianza en...
Leer más
En México durante la pandemia han predominado dos variantes: B.1.1.519 y delta

En México aún hay un gran número de casos de contagio, lo que indica que la pandemia no está controlada y la transmisión de la enfermedad se da a una tasa alta, por lo que es necesario vigilar al virus, “pues las mutaciones le van confiriendo propiedades”, señaló el Alejandro Sánchez Flores, Ph. D., responsable de la Unidad Universitaria de Secuenciación Masiva y Bioinformática del Laboratorio Nacional de Apoyo Tecnológico a las Ciencias Genómicas (LNATCG) del Consejo Nacional de Ciencia y Tecnología (CONACyT).[1]
¿Cómo enfrentar a este enemigo que parece invisible, que cambia de estrategia y plantea nuevos desafíos al personal médico y científico? En México, a raíz de la pandemia de COVID-19, surgió el Consorcio Mexicano de Vigilancia Genómica (CoviGen-Mex), un proyecto cuyo objetivo es investigar la evolución del virus en el país y en particular seguir la distribución y la aparición de nuevas variantes que pueden representar riesgos más allá del virus original.

Carlos Arias
El investigador Carlos Arias, Ph. D., Coordinador General del CoviGen-Mex, explicó a través del ciclo Lecciones y acciones a más de un año de la pandemia COVID-19, organizado por el Colegio Nacional, que un programa de secuenciación genómica mantenido a largo plazo puede tener un alto impacto en la salud pública, razón por la cual desde el inicio de la epidemia en México, como parte de la pandemia mundial, el Instituto de Diagnóstico y Referencia Epidemiológicos (InDRE) de la Secretaría de Salud ha dado seguimiento molecular genético a la presencia de diferentes virus en el país. Sin embargo, considerando el costo que tiene este tipo de metodología y el sofisticado equipo que se requiere, además del tiempo que demanda, era importante que tuviera apoyo de otras instituciones.
En México hay cuando menos 20 instituciones que contribuyen a secuenciar el virus, entre las que destacan el Instituto Nacional de Medicina Genómica (Inmegen), la Secretaría de Marina, la Universidad Autónoma de Nuevo León (UANL) y la Universidad Autónoma de Guerrero (UAGro).
“Dentro de este esquema y viendo la necesidad de tener un número mayor de secuencias virales que pudieran dar información más confiable, en enero de 2021 se creó el Consorcio Mexicano de Vigilancia Genómica (CoviGen-Mex)”. Este consorcio, al igual que las otras instituciones, envía las secuencias que se generan al Instituto de Diagnóstico y Referencia Epidemiológicos, como un laboratorio nacional de referencia, que a su vez las reporta a la Dirección General de Epidemiologia (DGE) y cuando es necesario, a la Organización Panamericana de la Salud (OPS) y a la Organización Mundial de la Salud (OMS).
Identificación de las variantes en México
La investigadora Celia Boukadida, Ph. D., del Centro de Investigación de Enfermedades Infecciosas (CISEI) y parte del consorcio, explicó que a lo largo del tiempo en México ha habido dos variantes predominantes que representan 80% de los casos: B.1.1.519, asociada a la segunda ola de aumento de personas infectadas en el país y ahora la variante delta, que ha disparado la tercera ola de infecciones en México.
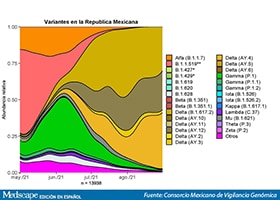
(Ampliar Imagen)
“Se identificó que la variante B.1.1.519 que se detectó en México en noviembre de 2020 llegó a tener una presencia de 76,6% en marzo de 2021, mientras que la variante alfa, detectada en enero de 2021, en mayo de este año representó 20,4% de los contagios. La variante gamma se detectó a partir de marzo de 2021 y se presentó en junio en 31,8%, en tanto que la variante delta apareció en febrero de 2021 y en agosto representó 95,7% de los virus detectados en México, siendo la variante que domina en el mundo actualmente”.
La estructura del virus que nos ocupa y sus propiedades biológicas
Arias recordó que el virus está formado por un genoma de ARN rodeado de una núcleoproteína y este complejo está protegido por una capa de lípidos derivado de la membrana celular, la cual está modificada por tres proteínas: envoltura, membrana y espícula, esta última la más relevante del virus en términos de las primeras interacciones que tiene la partícula viral con la célula hospedera, ya que es la que media la interacción del virus con el receptor, por lo que anticuerpos que van dirigidos contra ellos tienen la capacidad de ser neutralizantes, además es el antígeno que se ha usado hasta ahora en todas las vacunas aprobadas y en uso.
Asimismo, el genoma está formado por 30.000 nucleótidos; “se trata del más grande dentro de los virus de ARN, por ejemplo, el dengue cuenta con 11.000 nucleótidos, la influenza con 13.500 y el virus de inmunodeficiencia humana con 10.000. Durante la replicación del virus y del genoma viral como tal, la ARN polimerasa, encargada de hacer copias del genoma, introduce errores (mutaciones), que al tener un gran tamaño de genoma provoca muchas mutaciones, lo que podría llevar a que la progenie viral pudiera tener muchas mutaciones y causar que estas no fueran viables”.
Agregó que estos son los únicos virus de ARN que han desarrollado evolutivamente una maquinaria para corregir parte de estos errores. “Cuando la ARN polimerasa incorpora un nucleótido incorrecto, hay una actividad de exonucleasa que quita ese nucleótido incorrecto y añade al que corresponde, a pesar de eso, el genoma del SARS-CoV-2 y de los coronavirus en general tiene una tasa de evolución de 25 mutaciones por genoma por año”.
Esto quiere decir que si comparamos la secuencia del virus original detectado en enero de 2020 en Wuhan, China y lo comparamos con la secuencia de uno en 2021, lo que tendría este nuevo genoma son 25 mutaciones con relación al de un año antes. Estas mutaciones pueden ser puntuales (errores en la incorporación de nucleótidos), pero también pueden ser inserciones y deleciones de regiones del genoma o por eventos de recombinación involucrados en la evolución del genoma viral, ya que el virus evoluciona como resultado de la selección natural de aquellas mutaciones en su genoma que le confieren características favorables, como una mejor replicación, transmisión o evasión de la respuesta inmune.
“La variante delta tiene 53 mutaciones, en comparación con el virus original de Wuhan; son 53 diferencias de los casi 30.000 nucleótidos del sistema. De esas dos variantes, la original y la delta son idénticas en 99,82%”, añadió Boukadida.
Es importante mencionar que de acuerdo con esta tasa de error, usualmente se tiene una larga colección de virus con mutaciones que son dañinas para el virus, por lo que pueden hacer que no se replique o que no tengan mayor relevancia ni a favor ni en contra. “Estas mutaciones han ocurrido desde el inicio de la pandemia y son parte normal de la evolución de todos los virus”, señaló Arias.
¿Por qué seguir apoyando grupos de investigación en la materia?
Arias explicó que estos cambios presentes en diferentes niveles del genoma viral son una huella genética que permite identificar y seguir la dispersión de las variantes virales. “En general la epidemiología genómica ha permitido la identificación y dispersión de las variantes virales a través del mundo. En la actualidad se han secuenciado cerca de 3’415.000 virus en todo el mundo”. Esta es una cantidad sin precedente y una muestra de las mejoras tecnológicas que se han tenido a lo largo del tiempo.